Welcome!

We are a computational biology group at the
Meakins-Christie Laboratories of the McGill University School of Medicine.
Our lab focuses on studying cell dynamics in various biological processes in many diseases (e.g., developmental disorder, pulmonary diseases, cancers).
Decoding cell dynamics is essential for understanding the pathogenesis of diseases and finding novel therapeutics.
The existence of enormous heterogeneity in those diseases makes it challenging to decipher the unknown.
The advancing single-cell technologies that profile individual cell states provide unprecedented opportunities to tackle this problem,
which could drive biological discoveries and medical innovations in various fields (such as developmental and cancer biology).
However, the single-cell data presents numerous new challenges in developing computational models that bridge the biomedical data and potential discoveries.
Our primary research is to develop machine learning approaches (particularly probabilistic graphical models) to jointly analyze, model,
and visualize single-cell (and/or bulk) omics data (preferably longitudinal or spatial).
Such computational models will be used to help us derive a deeper understanding of the cell dynamics in different biological systems,
which will eventually benefit the public health with machine-learning driven new diagnostic and therapeutic strategies.
 DOLPHIN, a deep learning method that integrates exon-level and junction read data, representing genes as graph structures. These graphs are processed by a variational graph autoencoder to improve cell embeddings. DOLPHIN not only demonstrates superior performance in cell clustering, biomarker discovery, and alternative splicing detection but also provides a distinct capability to detect subtle transcriptomic differences at the exon level that are often masked in gene-level analyses. Please check here for details.
DOLPHIN, a deep learning method that integrates exon-level and junction read data, representing genes as graph structures. These graphs are processed by a variational graph autoencoder to improve cell embeddings. DOLPHIN not only demonstrates superior performance in cell clustering, biomarker discovery, and alternative splicing detection but also provides a distinct capability to detect subtle transcriptomic differences at the exon level that are often masked in gene-level analyses. Please check here for details.
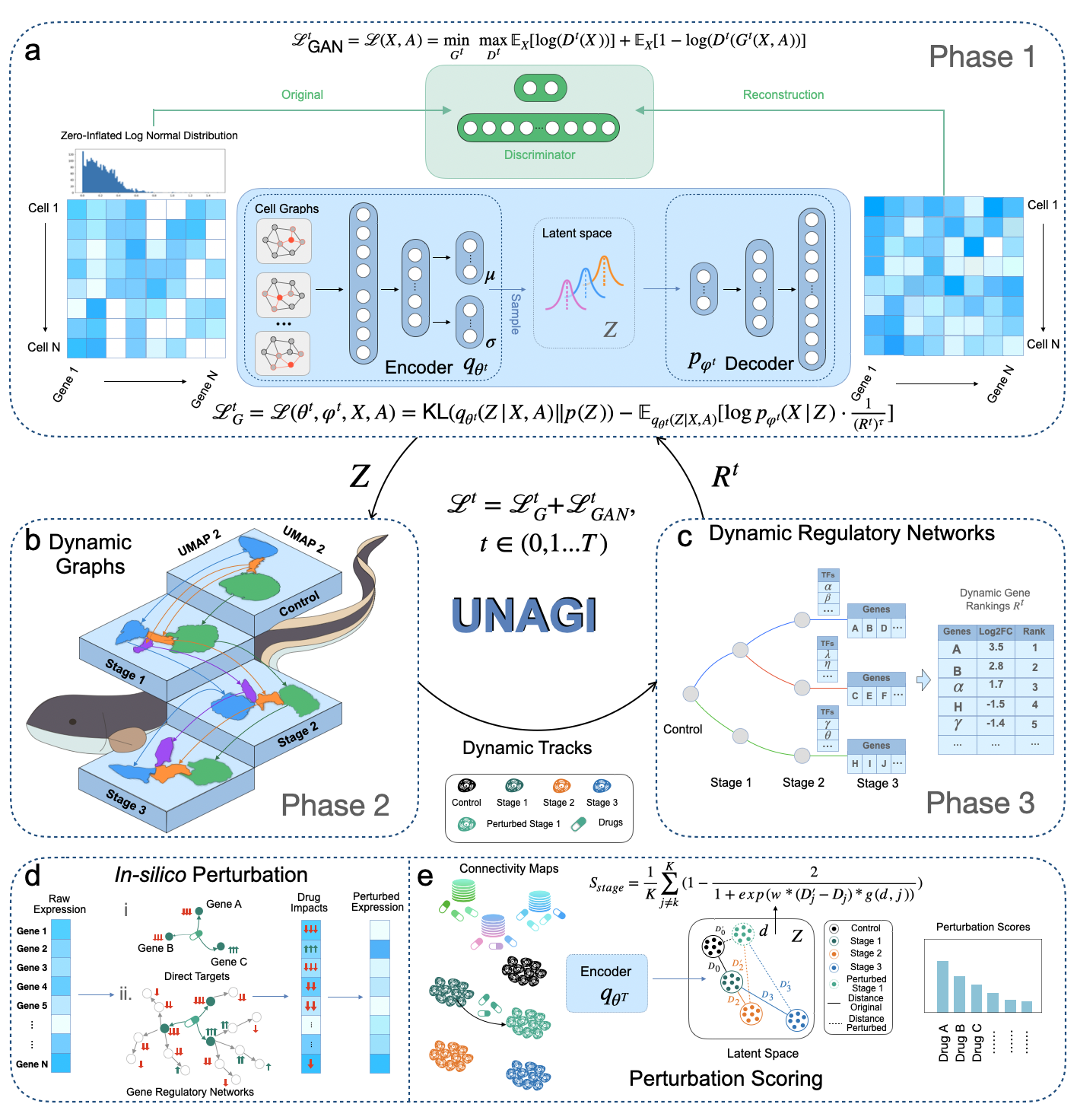 UNAGI is a comprehensive unsupervised in-silico cellular dynamics and drug discovery framework. UNAGI adeptly deciphers cellular dynamics from human disease time-series single-cell data and facilitates in-silico drug perturbations to earmark therapeutic targets and drugs potentially active against complex human diseases. Please check here for details.
UNAGI is a comprehensive unsupervised in-silico cellular dynamics and drug discovery framework. UNAGI adeptly deciphers cellular dynamics from human disease time-series single-cell data and facilitates in-silico drug perturbations to earmark therapeutic targets and drugs potentially active against complex human diseases. Please check here for details.
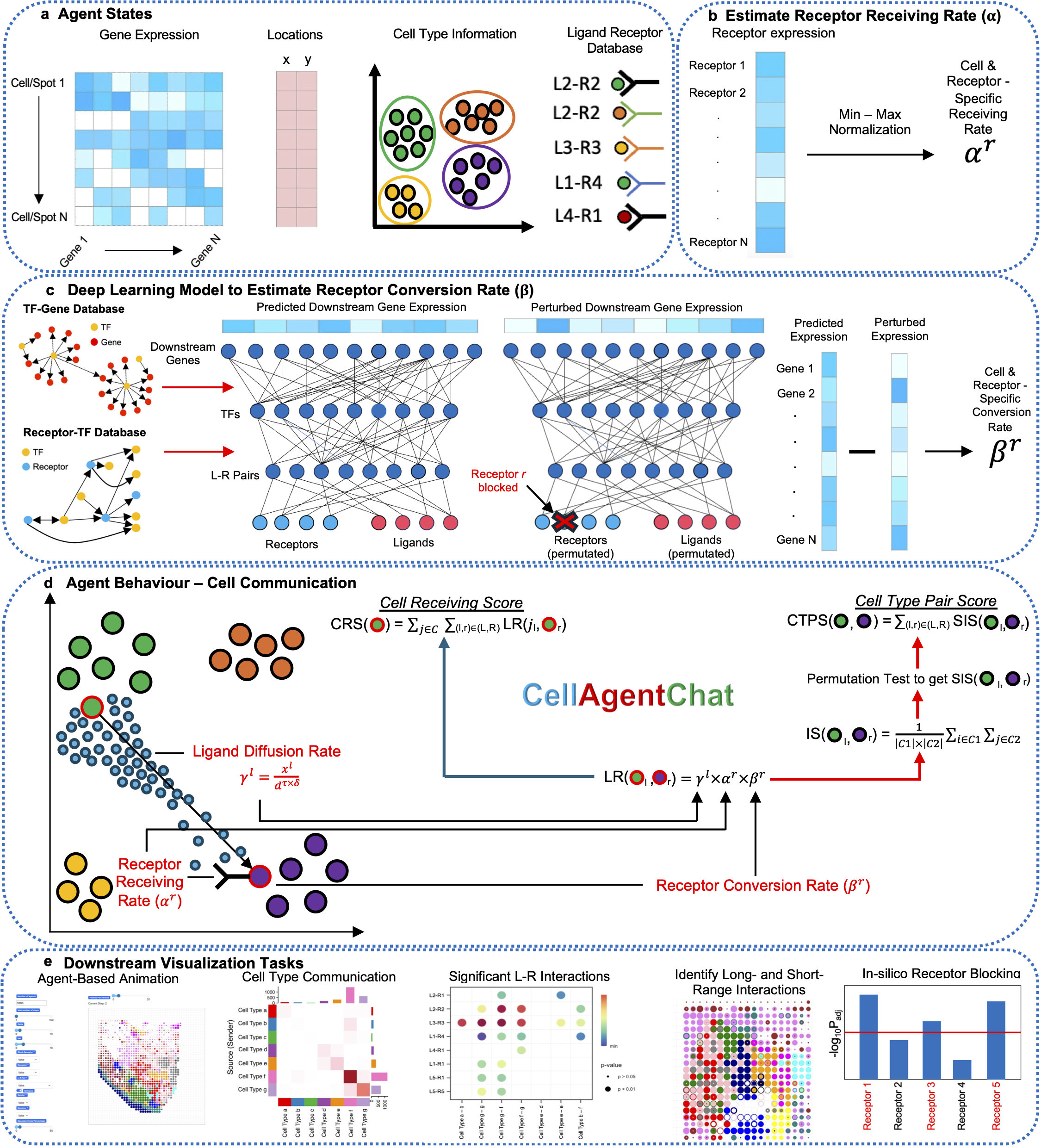 CellAgentChat constitutes a comprehensive framework integrating gene expression data and existing knowledge of signaling ligand-receptor interactions to compute the probabilities of cell-cell communication. Utilizing the principles of agent-based modeling (ABM), we characterize each cell agent through various attributes, including cell identities (e.g. cell type or clusters), gene expression profiles, ligand-receptor universe and spatial coordinates (optional). We quantify cellular interactions between sender and receiver cells based on the number of ligands secreted by the sender cells and subsequently received by the receiver cells. This process hinges upon three interrelated components: ligand diffusion rate (γl), receptor receiving rate (αr), and receptor conversion rate (βr). Please check here for details.
CellAgentChat constitutes a comprehensive framework integrating gene expression data and existing knowledge of signaling ligand-receptor interactions to compute the probabilities of cell-cell communication. Utilizing the principles of agent-based modeling (ABM), we characterize each cell agent through various attributes, including cell identities (e.g. cell type or clusters), gene expression profiles, ligand-receptor universe and spatial coordinates (optional). We quantify cellular interactions between sender and receiver cells based on the number of ligands secreted by the sender cells and subsequently received by the receiver cells. This process hinges upon three interrelated components: ligand diffusion rate (γl), receptor receiving rate (αr), and receptor conversion rate (βr). Please check here for details.
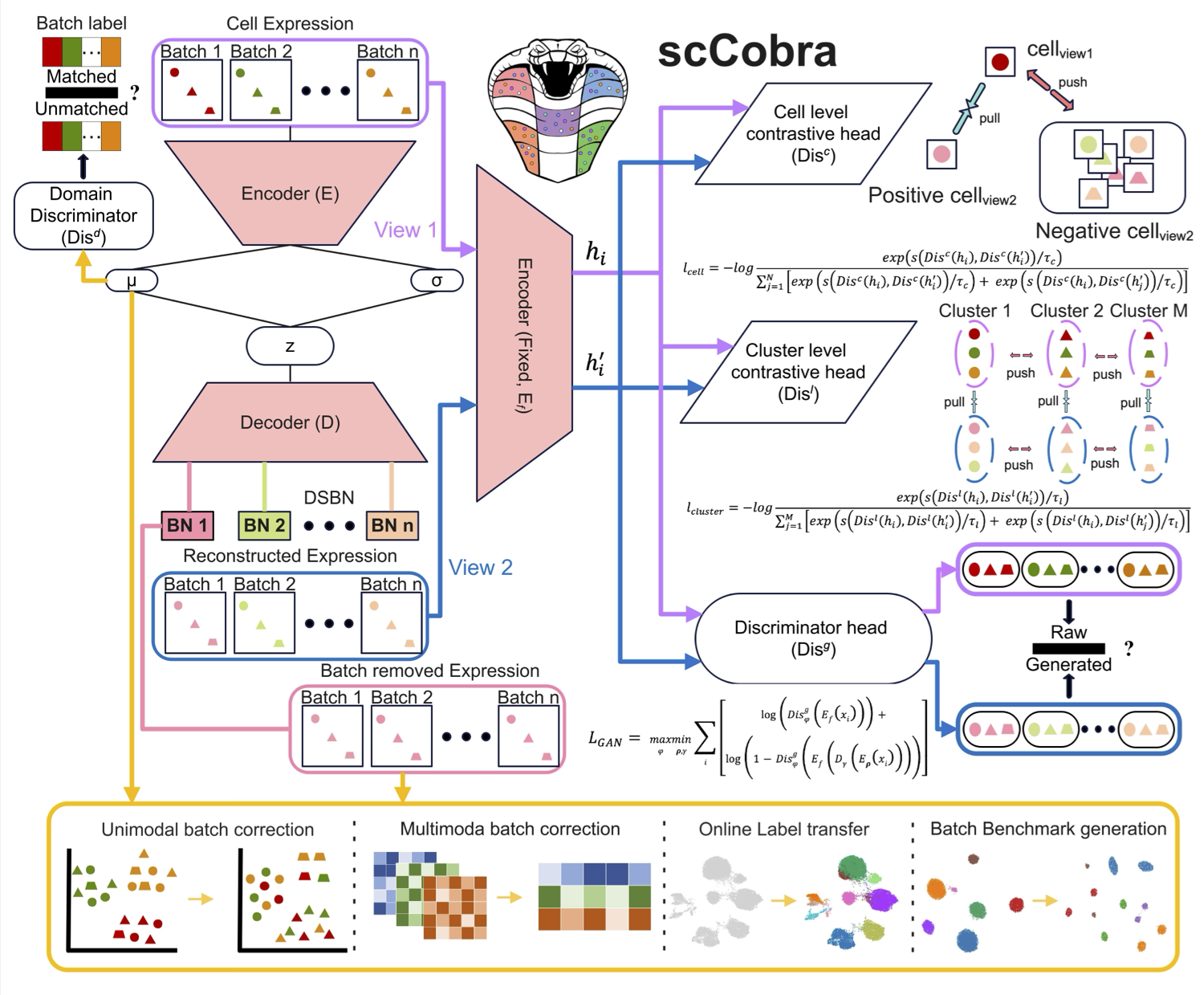 scCobra effectively mitigates batch effects, minimizes over-correction, and ensures biologically meaningful data integration without assuming specific gene expression distributions. It enables online label transfer across datasets with batch effects, allowing continuous integration of new data without retraining. Additionally, scCobra supports batch effect simulation, advanced multi-omic integration, and scalable processing of large datasets. By integrating and harmonizing datasets from similar studies, scCobra expands the available data for investigating specific biological problems, improving cross-study comparability, and revealing insights that may be obscured in isolated datasets. Please check here for details.
scCobra effectively mitigates batch effects, minimizes over-correction, and ensures biologically meaningful data integration without assuming specific gene expression distributions. It enables online label transfer across datasets with batch effects, allowing continuous integration of new data without retraining. Additionally, scCobra supports batch effect simulation, advanced multi-omic integration, and scalable processing of large datasets. By integrating and harmonizing datasets from similar studies, scCobra expands the available data for investigating specific biological problems, improving cross-study comparability, and revealing insights that may be obscured in isolated datasets. Please check here for details.
 MATES is a deep-learning approach that accurately allocates multi-mapping reads to specific loci of TEs, utilizing context from adjacent read alignments flanking the TE locus. This development facilitates the exploration of single-cell heterogeneity and gene regulation through the lens of TEs, offering an effective transposon quantification tool for the single-cell genomics community. Please check here for details.
MATES is a deep-learning approach that accurately allocates multi-mapping reads to specific loci of TEs, utilizing context from adjacent read alignments flanking the TE locus. This development facilitates the exploration of single-cell heterogeneity and gene regulation through the lens of TEs, offering an effective transposon quantification tool for the single-cell genomics community. Please check here for details.
 Single-cell multi-omics provides deep biological insights, but data scarcity and modality integration remain significant challenges. We introduce scCross, harnessing variational autoencoder and generative adversarial network (VAE-GAN) principles, meticulously designed to integrate diverse single-cell multi-omics data. Incorporating biological priors, scCross adeptly aligns modalities with enhanced relevance. Its standout feature is generating cross-modality single-cell data and in-silico perturbations, enabling deeper cellular state examinations and drug explorations. Applied to dual and triple-omics datasets, scCross maps data into a unified latent space, surpassing existing methods. By addressing data limitations and offering novel biological insights, scCross promises to advance single-cell research and therapeutic discovery.
Please read here for details.
Single-cell multi-omics provides deep biological insights, but data scarcity and modality integration remain significant challenges. We introduce scCross, harnessing variational autoencoder and generative adversarial network (VAE-GAN) principles, meticulously designed to integrate diverse single-cell multi-omics data. Incorporating biological priors, scCross adeptly aligns modalities with enhanced relevance. Its standout feature is generating cross-modality single-cell data and in-silico perturbations, enabling deeper cellular state examinations and drug explorations. Applied to dual and triple-omics datasets, scCross maps data into a unified latent space, surpassing existing methods. By addressing data limitations and offering novel biological insights, scCross promises to advance single-cell research and therapeutic discovery.
Please read here for details.
 scSemiProfiler is an innovative computational tool combining deep generative models and active learning to economically generate single-cell data for biological studies. It efficiently transforms bulk cohort data into detailed single-cell data using templates from selected representative samples. More details are in our manuscript.
scSemiProfiler is an innovative computational tool combining deep generative models and active learning to economically generate single-cell data for biological studies. It efficiently transforms bulk cohort data into detailed single-cell data using templates from selected representative samples. More details are in our manuscript.
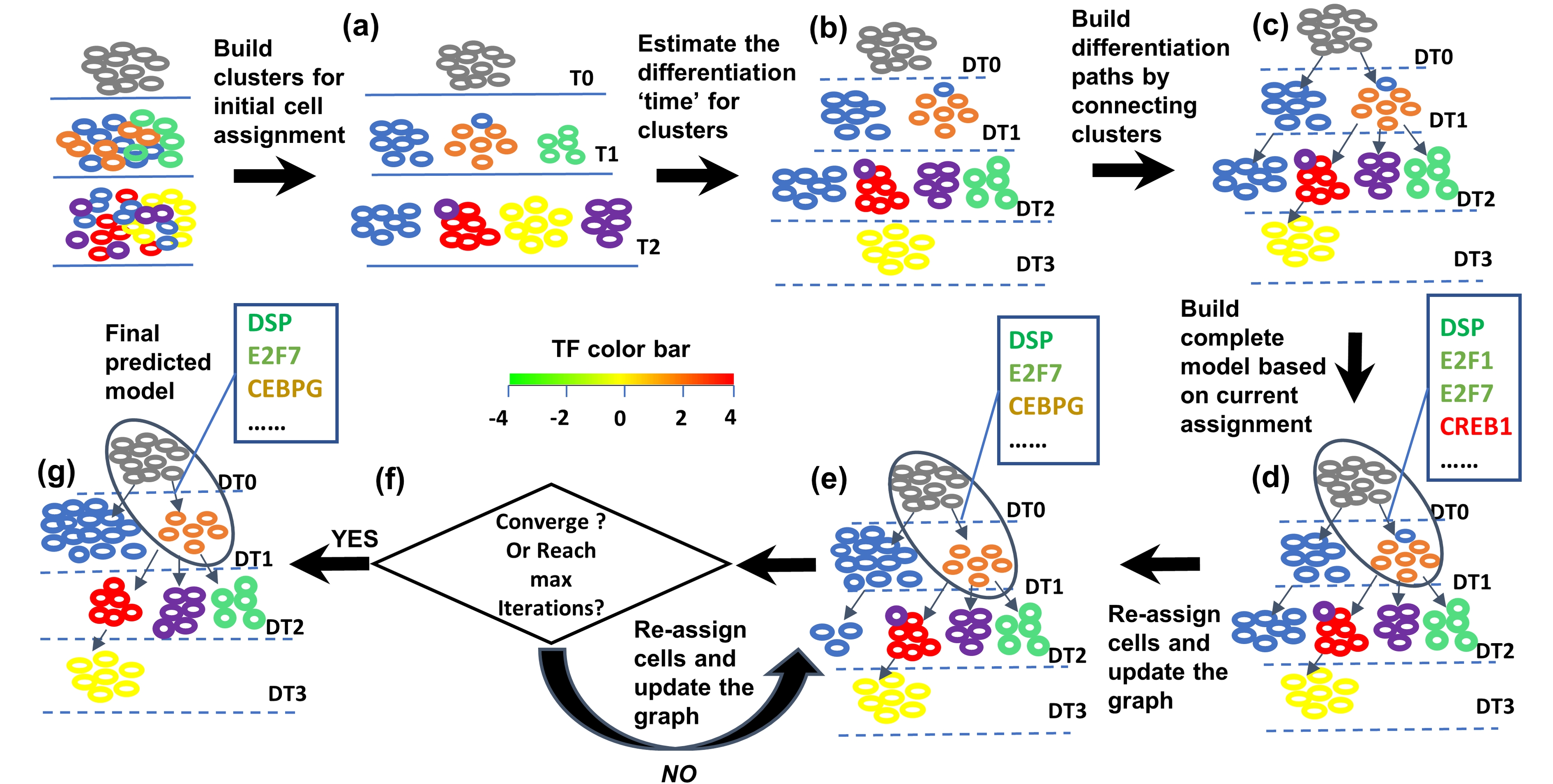 SCDIFF is a package written in python and javascript, designed to analyze the cell differentiation trajectories
using time-series single cell RNA-seq data. It is able to predict the transcription factors and differential genes associated
with the cell differentiation trajectories. It also visualizes the trajectories using an interactive tree-structure graph,
in which nodes represent different sub-population cells (clusters). Please check here for details.
SCDIFF is a package written in python and javascript, designed to analyze the cell differentiation trajectories
using time-series single cell RNA-seq data. It is able to predict the transcription factors and differential genes associated
with the cell differentiation trajectories. It also visualizes the trajectories using an interactive tree-structure graph,
in which nodes represent different sub-population cells (clusters). Please check here for details.
 Several recent studies focus on the inference of developmental and response trajectories from single cell RNA-Seq (scRNA-Seq) data. A number of computational methods,
often referred to as pseudo-time ordering, have been developed for this task. Recently, CRISPR has also been used to reconstruct lineage trees by inserting random mutations.
However, both approaches suffer from drawbacks that limit their use. Here we develop a method (named TBSP) to detect significant, cell type specific, sequence mutations from scRNA-Seq data.
We show that only a few mutations are enough for reconstructing good branching models. Integrating these mutations with expression data further improves the
accuracy of the reconstructed models. Please check here for details.
Several recent studies focus on the inference of developmental and response trajectories from single cell RNA-Seq (scRNA-Seq) data. A number of computational methods,
often referred to as pseudo-time ordering, have been developed for this task. Recently, CRISPR has also been used to reconstruct lineage trees by inserting random mutations.
However, both approaches suffer from drawbacks that limit their use. Here we develop a method (named TBSP) to detect significant, cell type specific, sequence mutations from scRNA-Seq data.
We show that only a few mutations are enough for reconstructing good branching models. Integrating these mutations with expression data further improves the
accuracy of the reconstructed models. Please check here for details.
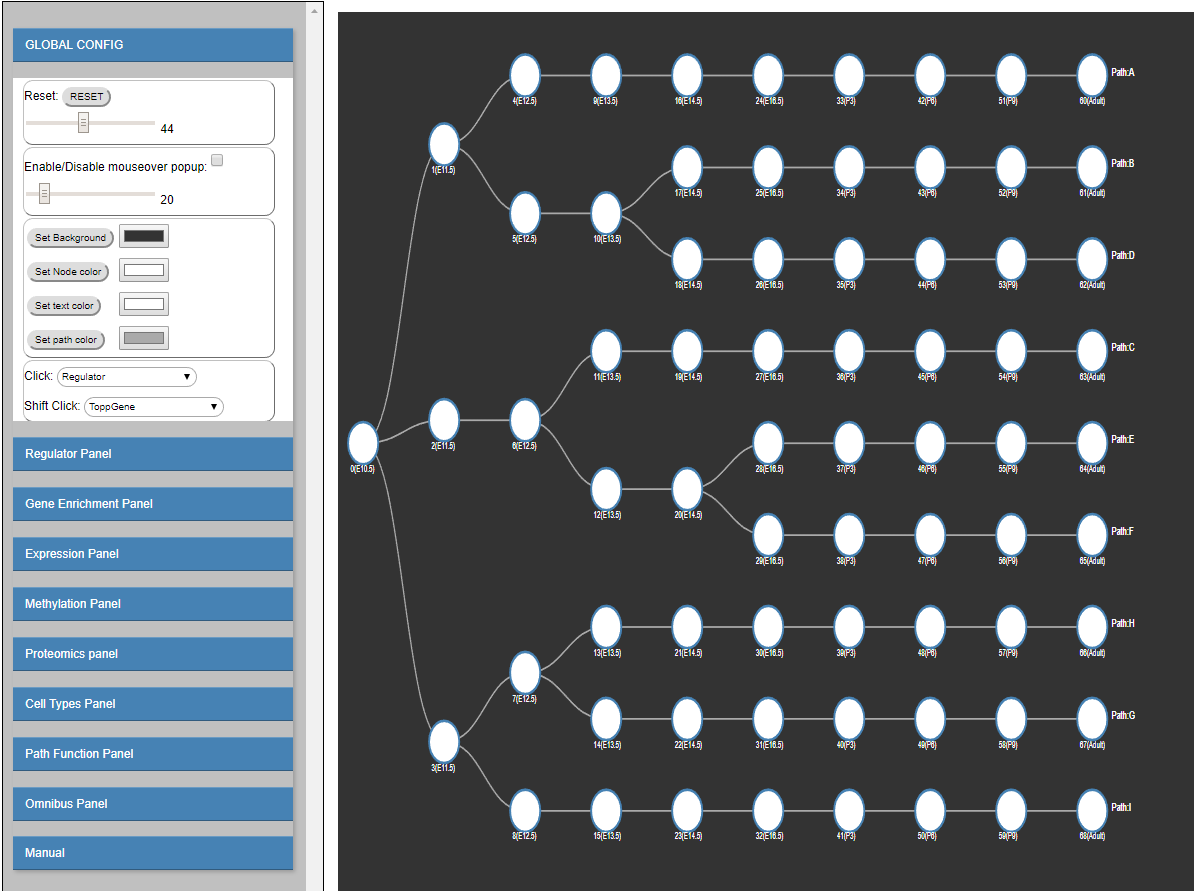 The Dynamic Regulatory Events Miner (DREM) software was initially developed to integrate static protein-DNA interaction data with time series
gene expression data for reconstructing dynamic regulatory networks. In recent years, several additional types of high-throughput time
series data have been used to study biological processes including time series miRNA expression, proteomics, epigenomics and single cell RNA-Seq.
Integrating all available time series and static datasets in a unified model remains an important challenge and goal.
To address this goal, and to enable interactive queries of the resulting learned models we have developed a new version of DREM termed interactive DREM (iDREM).
iDREM provides support for all data types mentioned above and more. Importantly, it also allows users to interactively visualize a gene, TF,
path or model-centric view of each of these data types, their interactions and their impact on the resulting model.
We showcase the functionality of the new tool by applying it to integrate several data types from multiple labs for modeling brain development regulatory networks.
Please read here for details.
The Dynamic Regulatory Events Miner (DREM) software was initially developed to integrate static protein-DNA interaction data with time series
gene expression data for reconstructing dynamic regulatory networks. In recent years, several additional types of high-throughput time
series data have been used to study biological processes including time series miRNA expression, proteomics, epigenomics and single cell RNA-Seq.
Integrating all available time series and static datasets in a unified model remains an important challenge and goal.
To address this goal, and to enable interactive queries of the resulting learned models we have developed a new version of DREM termed interactive DREM (iDREM).
iDREM provides support for all data types mentioned above and more. Importantly, it also allows users to interactively visualize a gene, TF,
path or model-centric view of each of these data types, their interactions and their impact on the resulting model.
We showcase the functionality of the new tool by applying it to integrate several data types from multiple labs for modeling brain development regulatory networks.
Please read here for details.
 The identification of microRNA (miRNA) target sites is fundamentally important for studying gene regulation.
There are dozens of computational methods available for miRNA target site prediction. Despite their existence,
we still cannot reliably identify miRNA target sites, partially due to our limited understanding of the characteristics of miRNA target sites.
The recently published CLASH (cross-linking ligation and sequencing of hybrids) data provide an unprecedented opportunity to study the
characteristics of miRNA target sites and improve miRNA target site prediction methods. Applying four different machine learning approaches to the CLASH data,
we identified seven new features of miRNA target sites. Combining these new features with those commonly used by existing miRNA target prediction algorithms,
we developed an approach called TarPmiR for miRNA target site prediction. Testing on two human and one mouse non-CLASH datasets,
we showed that TarPmiR predicted more than 74.2 % of true miRNA target sites in each dataset. Compared with three existing approaches,
we demonstrated that TarPmiR is superior to these existing approaches in terms of better recall and better precision.
Please read here for details.
The identification of microRNA (miRNA) target sites is fundamentally important for studying gene regulation.
There are dozens of computational methods available for miRNA target site prediction. Despite their existence,
we still cannot reliably identify miRNA target sites, partially due to our limited understanding of the characteristics of miRNA target sites.
The recently published CLASH (cross-linking ligation and sequencing of hybrids) data provide an unprecedented opportunity to study the
characteristics of miRNA target sites and improve miRNA target site prediction methods. Applying four different machine learning approaches to the CLASH data,
we identified seven new features of miRNA target sites. Combining these new features with those commonly used by existing miRNA target prediction algorithms,
we developed an approach called TarPmiR for miRNA target site prediction. Testing on two human and one mouse non-CLASH datasets,
we showed that TarPmiR predicted more than 74.2 % of true miRNA target sites in each dataset. Compared with three existing approaches,
we demonstrated that TarPmiR is superior to these existing approaches in terms of better recall and better precision.
Please read here for details.
❮
❯